















 203 320
203 320

end-joining predominates over homologous recombina-
tion, error-free repair of double-strand DNA breaks (DSBs).
Indeed, AR signaling may contribute to the formation of
DSBs at transcriptional hubs, also favoring non-homolo-
gous end-joining erroneous repair
[11] .Finally, these studies should provide the impetus and
hope that novel therapeutic strategies (beyond inhibition of
androgen synthesis and targeting the AR C-terminal LBD)
that inhibit oncogenic signaling driven by AR aberrations
may further transform the treatment of mCRPC
( Fig. 1 ).
Novel therapies that inhibit AR synthesis, mRNA splicing,
AR co-regulators, and the AR N-terminal domain have the
potential to overcome hormone-independent AR oncogenic
signaling driven by AR resistance aberrations. Although
therapeutically challenging owing to its lack of enzyme
activity and intrinsically unstructured nature, targeting of
the AR N-terminal domain, because of its presence in
implicated AR aberrations, is an exciting way forward. Some
of these hypotheses are currently being tested with
multiple agents including EPI-506 and TAS3681 in first-
in-human trials in mCRPC patients who may have
progressed on abiraterone and/or enzalutamide
(NCT02606123; NCT02566772). We envision that novel
drugs that target these aberrant AR splice variants will show
antitumor activity and have the potential to further
improve the treatment of advanced prostate cancer.
Conflicts of interest:
All of the authors are employees of The Institute of
Cancer Research, which has a commercial interest in abiraterone.
Johann S. de Bono has served as a consultant/advisory board member
for Astellas Pharma, AstraZeneca, Bayer, Genmab, Genentech, Glax-
oSmithKline, Janssen, Medivation, Orion Pharma, Pfizer, Tesaro, and
Sanofi.
Acknowledgments:
The authors acknowledge funding from Prostate
Cancer UK, the Prostate Cancer Foundation, Movember, Stand Up To
Cancer, the US Department of Defense, Cancer Research UK, the UK
Department of Health, the Academy of Medical Sciences, and
NHS funding to the NIHR Biomedical Research Centre at the Royal
Marsden and The Institute of Cancer Research. Joaquin Mateo is
supported by a Prostate Cancer Foundation Young Investigator
Award and a Prostate Cancer UK–Medical Research Council Fellow-
ship. Adam Sharp is supported by
[12_TD$DIFF]
a Medical Research Council
[13_TD$DIFF]
Fellowship.
References
[1]
De Laere B, van Dam P-J,[16_TD$DIFF]
Whitington T, et al. Comprehensive profiling of the androgen receptor in liquid biopsies from castra- tion-resistant prostate cancer reveals novel intra-AR structural variation and splice variant expression patterns. Eur Urol 2017;72:192–200.
[2] Henzler C, Li Y, Yang R, et al. Truncation and constitutive activation
of the androgen receptor by diverse genomic rearrangements in
prostate cancer. Nat Commun 2016;7:13668
. http://dx.doi.org/10. 1038/ncomms13668 .[(Fig._1)TD$FIG]
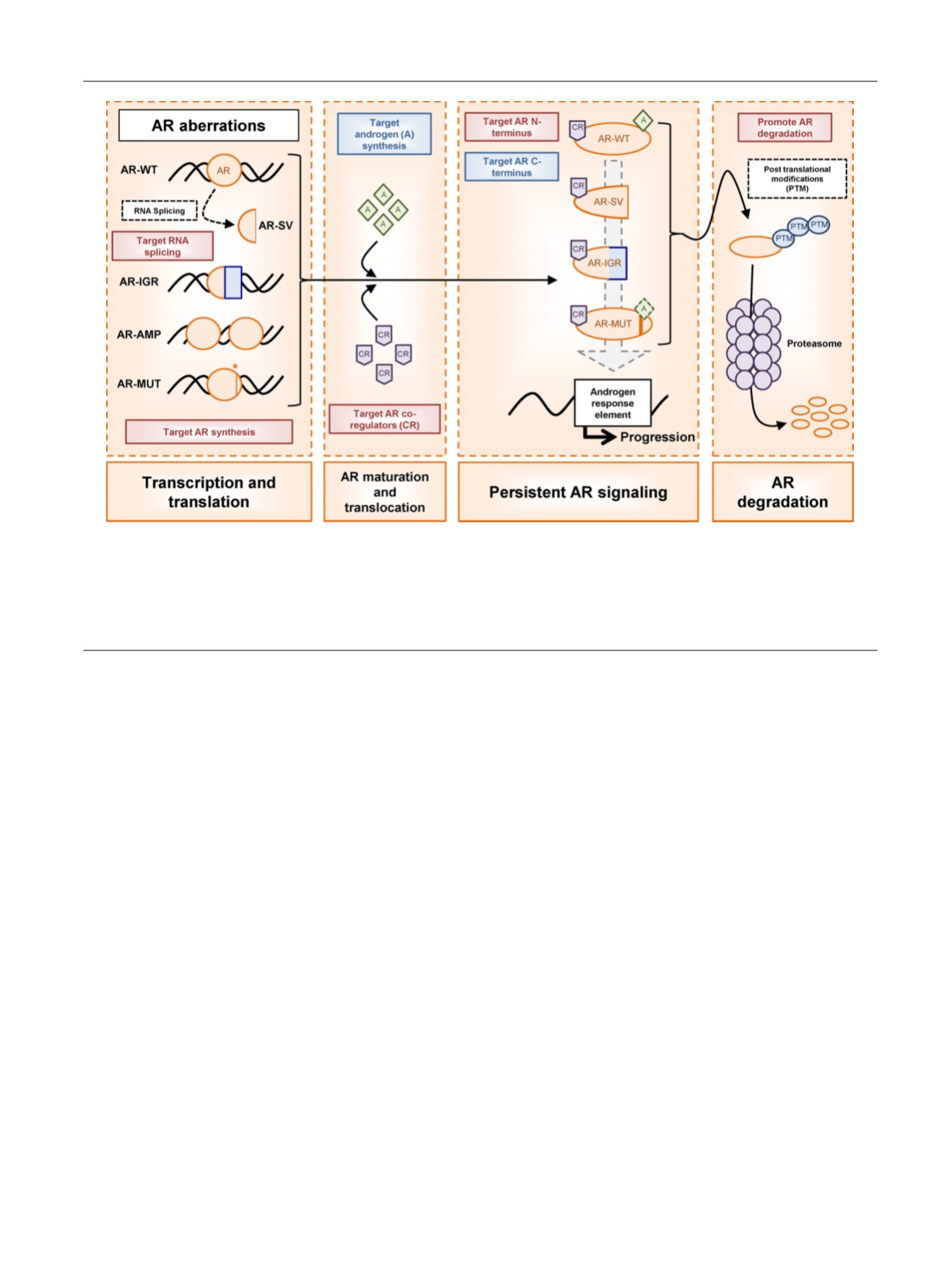
Fig. 1 – Novel therapeutic strategies to overcome androgen receptor aberrations in castration-resistant prostate cancer. Aberrations of the wild type
androgen receptor (AR-WT) including amplification (AR-AMP), gain-of-function mutations (AR-MUT), constitutively active splice variants (AR-SV), and
intra-genomic rearrangements (AR-IGR) lead to persistent AR signaling in castration-resistant prostate cancer (CRPC) despite androgen deprivation.
Therapies that target androgen (A) synthesis and the AR C-terminus are unlikely to overcome resistance mediated by AR aberrations in CRPC, as many
of these do not require androgen-dependent transactivation (blue shading). Novel therapeutic strategies targeting AR synthesis, RNA splicing, AR co-
regulators (CR), and the AR N-terminus, as well as those promoting AR degradation, have the promise to inhibit androgen-independent AR oncogenic
signaling driven through AR aberrations in CRPC (red shading).
E U R O P E A N U R O L O G Y 7 2 ( 2 0 1 7 ) 2 0 1 – 2 0 4
203